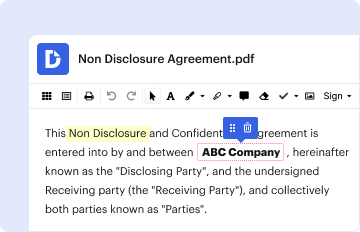
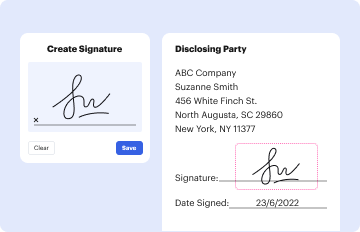
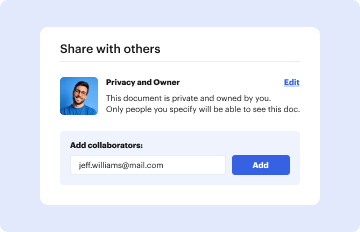

Start using our platform today to streamline your document editing and ensure a smooth submission process!
| Versions | Form popularity | Fillable & printable |
|---|---|---|
| 2015 | 4.7 Satisfied (36 Votes) |
At DocHub, your data security is our priority. We follow HIPAA, SOC2, GDPR, and other standards, so you can work on your documents with confidence.
Learn more







