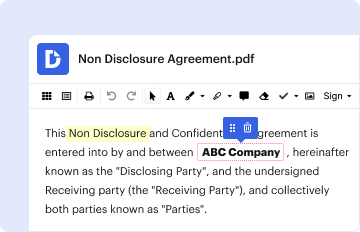
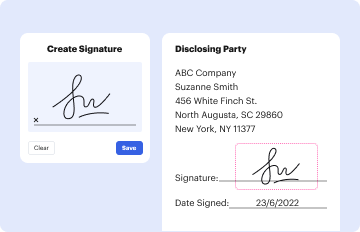
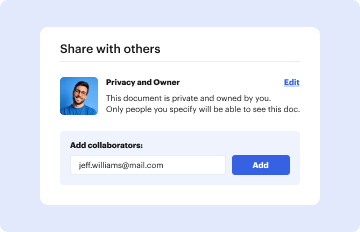
Type text, add images, blackout confidential details, add comments, highlights and more.
02. Sign it in a few clicks
Draw your signature, type it, upload its image, or use your mobile device as a signature pad.
03. Share your form with others
Send instructions fda via email, link, or fax. You can also download it, export it or print it out.
How to use or fill out form 3794 with our platform
Ease of Setup
DocHub User Ratings on G2
Ease of Use
DocHub User Ratings on G2
Click ‘Get Form’ to open Form FDA 3794 in the editor.
Begin by filling out the 'APPLICANT, HOLDER OR OWNER' section with the legal name and address of the entity submitting the application.
In the 'REPRESENTATIVE OR U.S. AGENT' field, provide contact details for a person authorized to respond to FDA inquiries, ensuring they are a U.S. agent if applicable.
Indicate the relevant 'FISCAL YEAR' for your submission, noting that it starts on October 1 of the previous calendar year.
Select the appropriate 'GENERIC DRUG USER FEE TYPE' by checking the corresponding box for ANDA, PAS, Type II API DMF, or backlog fee.
Complete sections related to ANDA/PAS information by entering application numbers and product names as required.
Fill in facility information including name, address, FEI number, and DUNS number as necessary.
Review all entries for accuracy before submitting your completed form through our platform.
Start using our platform today to streamline your form completion process for free!
Additional information may also include the Generic Drug User. Fee (GDUFA) Coversheet (Form FDA 3794) for GDUFA Master Files (Type II APIs), when applicable.Read more
Cookie consent notice
This site uses cookies to enhance site navigation and personalize your experience.
By using this site you agree to our use of cookies as described in our Privacy Notice.
You can modify your selections by visiting our Cookie and Advertising Notice.