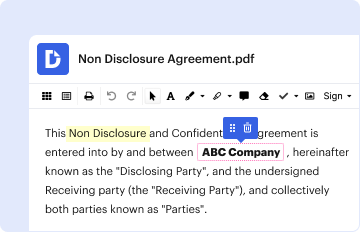
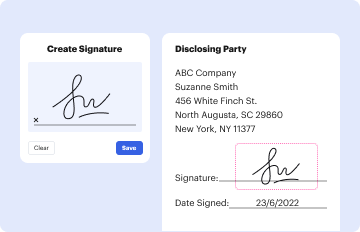
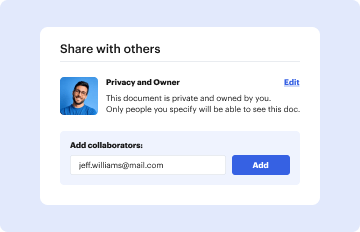

Start using our platform today to streamline your document editing and ensure compliance with FDA regulations!
At DocHub, your data security is our priority. We follow HIPAA, SOC2, GDPR, and other standards, so you can work on your documents with confidence.
Learn more